
热门:
180-3804-4554
185-7640-1396
登录


关于快可利
快可利实验室技术服务事业部以“专业、专注、专心地满足客户需求”为宗旨、对各领域、各类型的实验室都有丰富的设计规划、装修施工、技术培训、专业咨询经验。16年来,快可利实验室技术服务事业部已设计、装修、认可咨询和技术支持了500+企业,快可利实验室技术服务事业部将利用这些宝贵的经验帮助客户从实验室现场布局设计、装修施工、家具配置、设备咨询、项目验收、人员培训、体系建立、认证认可等多方面来实现实验室一站式解决方案。
联系我们  180-3804-4554
180-3804-4554






关于快可利
快可利实验室技术服务事业部以“专业、专注、专心地满足客户需求”为宗旨、对各领域、各类型的实验室都有丰富的设计规划、装修施工、技术培训、专业咨询经验。16年来,快可利实验室技术服务事业部已设计、装修、认可咨询和技术支持了500+企业,快可利实验室技术服务事业部将利用这些宝贵的经验帮助客户从实验室现场布局设计、装修施工、家具配置、设备咨询、项目验收、人员培训、体系建立、认证认可等多方面来实现实验室一站式解决方案。
联系我们 180-3804-4554
CTS新闻 CTS公告 行业动态 学术论坛

AABB认证咨询-AABB CT 标准设备管理解读:构建细胞治疗设备全生命周期合规体系
发布日期:2025-09-15 16:24:51
文章来源:
阅读次数:1
分享:






微信
贴吧
QQ空间
邮箱
在细胞治疗领域,设备是保障产品质量与患者安全的核心载体 —— 从细胞采集的离心机到储存用超低温冰箱,从制备环节的生物反应器到追溯用信息系统,任何设备的偏差都可能导致治疗失败或安全风险。AABB CT 标准 “设备” 章节(3.0-3.5 条款)围绕设备全生命周期构建了系统化管理框架,覆盖从采购前规划到应急替代的全流程,为机构提供了可落地的设备合规指南,对解决当前细胞治疗设备管理中 “重采购、轻维护”“信息系统安全不足” 等痛点具有关键指导意义。
一、设备全流程管控:从源头规范到唯一性标识
条款 3.0 与 F3.1 确立了设备管理的 “前置化” 原则,要求机构在设备采购阶段即建立管控逻辑。F3.1 明确的三大控制要素 —— 采购前规格确定、预期用途确认(含精密度 / 准确度)、唯一标识,是避免 “设备错配” 的核心防线。以细胞治疗常用的 “细胞计数仪” 为例,采购前需根据临床需求确定检测范围(如 10⁴-10⁷ cells/mL)、精度要求(CV≤5%),到货后需通过标准品验证其准确度,避免因设备性能不达标导致细胞剂量计算错误。而 “唯一标识” 要求(如设备编号 + 位置编码)则是后续追溯的基础,某 CAR-T 治疗中心通过赋予每台超低温冰箱唯一编码,成功在一次温度波动事件中快速定位涉事设备及储存的 23 份细胞产品,为应急处置争取了时间。

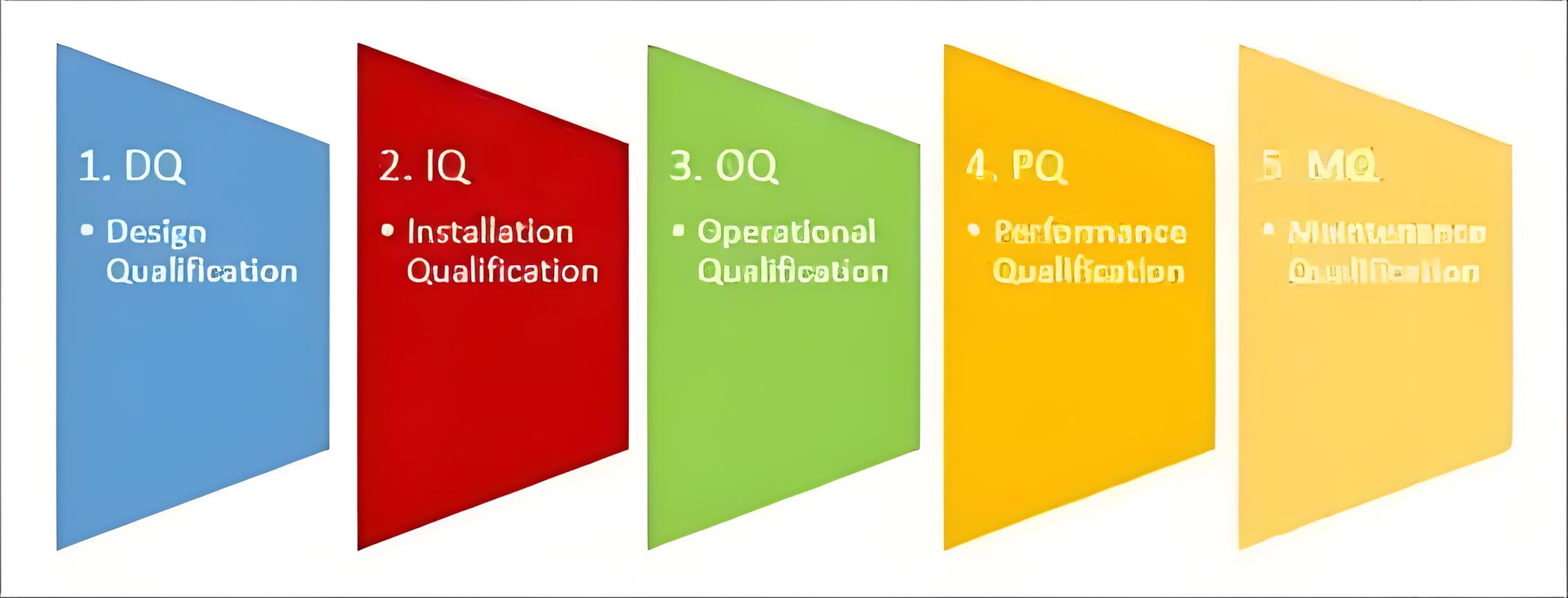
设备确认是确保 “设备能用、好用” 的关键环节。3.2 条款将确认分为安装确认(IQ)、运行确认(OQ)、性能确认(PQ)三阶段,形成递进式验证体系:安装确认(3.2.1)需对照制造商规范核查环境适配性(如生物安全柜需确认通风量、接地电阻);运行确认(3.2.2)需验证设备各组件功能(如细胞输注泵的流速调节范围、报警功能);性能确认(3.2.3)则需在实际使用场景下验证设备是否满足治疗需求(如冷冻干燥机对细胞存活率的影响)。这一流程与 GMP 设备验证逻辑一致,某干细胞库通过严格执行三阶段确认,使设备故障导致的产品损耗率从 12% 降至 3% 以下。
二、监控与维护:构建设备 “健康管理” 闭环
条款 3.3 聚焦设备使用中的持续管控,核心是通过定期维护与校准确保设备长期稳定。F3.3.1 从校准全流程提出要求:需明确校准参数(如 pH 计的校准点)、频率(如天平每月 1 次)、方法(需符合国家测量标准),并保护校准设定不被篡改。实践中,机构可建立 “设备校准台账”,标注每台设备的校准周期、责任人及历史记录,某血液中心通过该台账实现校准逾期率为 0。尤为关键的是,当设备超校准范围时(F3.3.2),需立即启动偏差评估 —— 不仅要检修设备,还要追溯该设备此前处理的细胞产品是否合格,如某机构发现一台细胞计数仪超差后,通过评估确认其此前检测的 15 份产品中 2 份剂量计算偏差,及时更换产品避免了临床风险。
设备的日常维护与环境管控同样重要。F3.3.3 要求制定清洁消毒流程(如细胞处理台每日用 75% 乙醇消毒)、故障通知机制(如设备报警后 10 分钟内通知维修人员),并由资质人员按制造商建议检修。以超低温冰箱为例,需每周检查液氮液位、每月清洁滤网,这些维护措施能将设备故障率降低 40% 以上;同时,环境条件需适配设备需求(如 PCR 仪需置于恒温恒湿房间),避免环境因素影响设备精度。

三、设备追踪:打通细胞治疗 “溯源链路”
F3.4 的设备追踪要求是实现细胞产品全生命周期可追溯的关键支撑。机构需保存设备使用记录,确保每一份细胞产品能关联到采集(如血细胞分离机)、处理(如生物反应器)、储存(如超低温冰箱)、分发(如冷链运输设备)全环节的设备信息。这种追踪能力在产品召回时尤为重要 —— 若某批次细胞产品因储存设备温度波动存在质量风险,通过设备追踪可快速定位同设备储存的所有产品,避免风险扩散。某国际细胞治疗机构的实践显示,完善的设备追踪系统可使产品召回响应时间缩短至 2 小时内。
四、信息系统与替代方案:应对数字化时代的设备风险
3.5 条款针对细胞治疗的数字化趋势,构建了信息系统的合规框架,涵盖 12 项核心要求,核心是 “数据完整性” 与 “系统安全性”。例如,要求系统具备数据验证功能(如输入细胞剂量时自动校验范围)、唯一身份标识(供者 / 产品 / 受者一一对应)、受保护健康信息(PHI)保密机制(如数据加密存储)。同时,人员需经培训合格方可使用系统,避免因操作失误导致数据错误。
3.5.1 的替代系统要求是应对 “数字中断” 的关键预案 —— 当电子系统故障时,需有纸质备份或备用系统确保关键活动不中断(如手工记录细胞储存温度),且 F3.5.1.1 要求定期测试替代系统(如每季度模拟系统宕机演练)。某 CAR-T 中心通过测试发现,其原替代系统的纸质记录表格存在信息缺失,及时完善后确保了应急场景下的数据完整性。
编辑:
地区:
(1).23e3596.png)
评论列表
 徐老师
徐老师  王老师
王老师  张小姐
张小姐  陈小姐
陈小姐  郭小姐
郭小姐 
.2f44860.png)
热线电话
 18576401396 18038044554
18576401396 18038044554 .36720e2.png)
在线咨询
.43b2c75.png)
转发网站
微信
贴吧
QQ空间
邮箱
.4f4ea8a.png)
在线留言
.a3a1a2a.png)
回到顶部
友情链接:
网站备案号:粤ICP备2020125901号 版权所有:深圳市快可利信息技术有限公司
电话:180-3804-4554 传真:0755-89335196 地址:深圳市龙岗区富安大道华耀城12栋909